EGEE 510
PHYSICAL CHEMISTRY IN ENERGY,
GEO-ENVIRONMENTAL AND MINERAL ENGINEERING
Here is
the Fall 2008 syllabus!
In 1927 Heitler and
That's why physical chemistry is today THE most important
scientific 'discipline' that forms the basis of ALL (OK... maybe not all, but
certainly MOST) engineering applications.
In most practical situations,
chemical kinetics (see Chs. 21-24 of Atkins) is still an experimental field,
although we can increasingly rely on order-of-magnitude estimations using first
principles. In the first part of our course we shall review the highlights of
phase and chemical equilibrium which allows us to determine the concentrations
of species of interest and the ultimate composition of any
reacting mixture. In the second part we shall review the highlights of chemical
kinetics -- which allow us to determine more realistic compositions --
and illustrate their relevance and applicability to selected energy,
geo-environmental and mineral engineering issues.
The objective of this
course, apart from reviewing the applications of thermodynamics and kinetics,
is to provide an exercise in targeted and stimulated self-study. In particular, the key to our success will be
that you come to 'class' prepared to discuss the topics and issues
summarized below (and discussed at length in a physical chemistry textbook).
--OVERVIEW AND APPLICATIONS OF
THERMODYNAMICS--
As an introductory exercise, during
the first week of class, when we shall have no group meetings (i.e.,
we shall have no “class”), you are asked to do the following:
(1) Develop a habit of
reading and learning with the following tools beside you: (a) Internet (e.g.,
google.com, Web of Science); (b) Excel, and (c) Mathematica (or any other
higher-power math software than Excel, especially for quick visualization of
equations).
(2) Retrieve from the
electronic library the classical JACS paper by Brunauer, Emmett and Teller
where the BET equation has been presented for the first time. (Do you need help to find this
paper?)
(3) Which phase (or
chemical?!) equilibrium does this equation describe?
(4) If possible, based on the
information provided, use Excel to obtain the surface area of any material
discussed in the paper. Here is some
material for discussion...
(5) Analyze some of the
tables and figures as carefully as you can, and try to ‘derive’ (or identify)
some of the numbers that appear in some of them, based on the information
provided in the text or in (an)other table(s) or figure(s).
(5a) See here a few
relevant Mathematica calculations... And
here an
update... Be prepared to discuss them (or to ask specific questions about them)!
(6) Use the “LeChatelier
principle” to justify the adsorption trends shown in Table IV. (Is adsorption
always exothermic? Does it matter whether it’s ‘physical’ or ‘chemical’?)
Some of the principal milestones in the development of thermo:
-1760s: Black, heat capacities, latent heats
(“calorimetry”)
-1840s: Mayer, interconversion of heat and work (1st
law)
-1840s: Joule, interconversion of heat and work (Who’s done
it first... A fascinating story!)
-1850s: Kelvin, absolute temperature and 2nd law
-1850s: Clausius, entropy and 2nd law
-1870s: Gibbs, phase rule and chemical potential (“free
energy”)
-1880s: Helmholtz, equilibrium and free and bound energy
-1880s: van’t Hoff, equilibrium constant
-1900s: Nernst, 3rd law
Isn’t it remarkable that in
only half a century essentially all the key
concepts became rather clear (despite the fact that the relevant issues --
e.g., energy, heat, work -- had been studied for centuries)?!
Key G/S
phase equilibrium concepts:
-collision frequency (units?)
-surface coverage (monolayer, multilayer)
-intermolecular interactions
-adsorption isotherm (Langmuir, BET, Freundlich, Dubinin,
etc.)
-others?
HW1 (accepted until 9/17, preferably in
appropriate Angel dropbox), Atkins8: D7.1-7.5, E7.1, E7.9, E7.12, P7.2,
P7.4, A7.36, D25.3, D25.5-25.7, E25.1, E25.4, E25.8, E25.13, P25.2, P25.6,
A25.33. (Note: Parts of these problems
will be solved during our class discussions or as ‘hints’, to be posted here in
due course.)
Criteria for selection of a ‘good’ paper for (exam)
analysis:
-Any research paper (or thesis,
or report) must contain figures and/or tables that present one or all of the
following types of results:
(a) ‘Raw’ data: e.g., temperature or pressure vs.
composition.
(b) Data analysis: in our case, this is typically
the determination of equilibrium parameters (e.g., properties such as
equilibrium constant or partition coefficient) for a system, reaction, or
process.
(c) Correlations: establishment (e.g., discovery,
confirmation, rediscovery) of relationships between structure and properties
(in a ‘scientific’ study) or between properties and behavior (in an
‘engineering’ study).
-A ‘good’ paper for analysis
will contain information in each one, or at least two, of these categories.
HW1 hints/solutions:
-In order to use Sandler’s chemeq.bas program, do you agree
that the following line should be added for CuSO4 to the react.dta
file? (And for CuSO4*5H2O?)
“CuSO4”,-661.8,-771.4,0.414,-26.7,-22.6,72.2,0.
-Here
is a partial solution to A7.36. I suggest you use it
as a ‘template’ for the solution of the other problems, in terms of (a) results
provided, (b) assumptions made and (c) commentary offered. Be sure to
explicitly address ALL the questions posed in the statement of the problem.
-E25.1(a): Hydrogen(i)=1.08x1025 m-2
s-1; Hydrogen(ii)=1.44x1018 m-2 s-1;
Propane(i)=2.3x1024 m-2 s-1,
Propane(ii)=3.1x1017 m-2 s-1.
-E25.4(a): 12.7 m2/g.
-E25.8(a): (i) 0.21 kPa;
(ii) 22 kPa.
-E25.13(a): -12.3 kJ/mol.
-P25.2: (a) ca. 2x108;
(b) ca. 2x103.
-P25.6: Assuming 0.157 nm2 per H2
molecule (reasonable?), the Langmuir eqn gives a surface area of ca. 6 m2/g.
-A25.33: (a) R2=0.988 for a, R2=0.932
for b (good enough?); (b) ka=3.65x10-3 kg/kg, kb=2.7x10-5
ppm-1, DHa=-8.69 kJ/mol, DHb=-15.6 kJ/mol; (c) Does the cited paper shed any light on
this issue? (See also this summary
and this
analysis of the Langmuir equation.)
-E7.1(a): 2.85x10-6, 240 kJ/mol, 0 kJ/mol.
-E7.9(a): yB=0.90, yIB=0.10. In
E7.9(b) note the error regarding the number of moles of N2, so the correct
solution is yNO=0.0176.
-E7.12(a): Above approx. 1150 K. For E7.12(b) the calculation
shown assumes that Cp is independent of T; it’s not easy to find
Cp=f(T) for the hydrate, but if the 298 K value is used, the calculated
decomposition temperature (ca. 400 K) is very close to that shown in the
solution manual (397 K).
-P7.2: (a) 1.24x10-9; (b) 1.29x10-8;
(c) 1.8x10-4.
-P7.4: (a) K = 1.22x10-6 (1395 K), 2.80x10-6
(1443 K), 7.23x10-6 (1498 K). This is in reasonable agreement with
Chemeq: 9.3x10-7, 2.1x10-6, 4.9x10-6. It’s
much better to check the reliability of Chemeq, and then use it to determine
the other thermochemical parameters, than to make all these calculations by
hand.
Discussion questions:
7.1: How does the mixing of
reactants and products affect the position of chemical equilibrium? (Does the
equilibrium constant depend on the concentrations of the species involved in
the reaction?)
7.2: What are the various
ways in which K can be expressed as a ratio of ‘concentrations’?
7.3: Effects of pressure (for deltaN≠0) and T?
7.4: On the van’t Hoff plot, sketch a typical result
for an endothermic and an exothermic reaction. What are the ‘molecular’
implications (in terms of favoring reactants or products)? (Hint: A quick and
convenient use of Sandler’s chemeq.bas should be useful here.)
-Here is a
comprehensive van’t Hoff plot
available from the literature. Can you reproduce some of these graphs?
Summary of key issues in V/L equilibrium:
-see handout “Summary of VLE criteria”
-solubility (Henry’s law): G --> L
-vapor pressure: L --> G
-single component: Antoine
eqn, etc.
-solutions (mixtures): see Figure 6.8, Atkins8
-enthalpy of vaporization
(Clausius-Clapeyron eqn)
-phase rule and
phase
diagrams
-fugacity [f = (fugacity
coefficient)(pressure)] and activity [a = (activity coefficient)(mole fraction
in liquid)]
-standard
states (‘solvent’: Raoult’s law; ‘solute’: Henry’s law)
-ideal solutions (DHmix=0, HE=0)
-regular
solutions (DHmix≠0,
HE≠0, DSmix=0)
-calculations
using equation of state (e.g., vdW)
-here is a Mathematica file
that allows easy preparation of the p vs. composition diagram for VLE
-for analysis of combined phase and chemical equilibrium,
see handout “G/L Rxn Equilibrium”, as well as the accompanying Mathematica
file.
HW2 (due 10/1), Atkins8: D1.3-1.6, E1.13, E1.22, A1.25, A2.45, D3.6-3.8, P3.18, A3.39, E4.8,
P4.8, A4.22, A4.23, E5.3, E5.10, P5.9, P5.12, A5.25, D6.1, E6.2, E6.5, P6.3.
-the importance of having some math software is illustrated
here for
convenient differentiation (E5.10) and here for a quick
solution of integrals in eqn 3.60 (P3.18)
-E1.22a: b=4.6x10-5 m3/mol; Z= 0.661
(Makes sense? Why?)
-P3.18: Using a fourth-order polynomial (Why? Necessary?), f = 0.81, f = 81 atm.
-E4.8a: DHvap=49.1 kJ/mol; Tb=215
oC; DSvap=101 J/mol/K (Can check these values?)
-P4.8: 83.6 oC; DHvap=38.0
kJ/mol (Can check these values?)
-A4.22: Tb=111.5 oC; DHvap=7.9 kJ/mol (Can check these values?)
-E5.10a: n6/n7=1.0; m6/m7=0.86.
-P5.12: Check the Francesconi et al. (1996) reference!
-E6.2a: P=58.6 kPa, xA=0.27.
-E6.5a: (i) yM = 0.36; (ii) yM = 0.82.
-P6.3: Is Raoult's law (yiP=xiPi,sat)
mentioned? Applicable? (Here
is another example of the significance of Raoult's law and deviations from it.)
Note: Our next group meeting will be on 9/29. In the
meantime, be sure to verify the partial solutions and/or hints for the HW
problems that will be posted here. Also, see whether you can find a suitable
research paper that is of interest to you and contains figures and/or tables
that illustrate phase and/or chemical equilibrium issues. Upon mutual
agreement, such a paper will probably serve as the basis for your Exam1. Of
course, for help with any of these activities, as well as further clarification
of the material posted on the class web site, be sure to contact me by e-mail
24/7.
Similar tools and concepts
can be extended, in a more or less straightforward way, to understand L/S
phase and chemical equilibria... but this must be left for your own
study (when needed). HW3 illustrates a few examples. It is also meant to cover,
albeit very briefly, the increasingly important field of statistical thermodynamics (Chs.
16 and 17 in Atkins8), which, among other virtues, forms the basis for quantum
thermochemistry calculations -- e.g., vibrational frequencies and energy
changes between reactants, products and their transitions states (see, for
example, Section 11.8 in Atkins8) -- and thus provides a natural link to our
next topic of discussion, chemical kinetics.
Questions to be explored
whenever you read a paper that is important for your research: (And for your
EGEE 510 exams?)
(i)
Which equation(s)
in this paper are identical, or similar, to equation(s) in Atkins? Does the
paper analyze phase equilibrium or chemical equilibrium, or both?
(ii)
Which graph(s) in
this paper are identical, or similar, to graph(s) in Atkins?
(iii)
Does the
information provided in the paper (e.g., tables) allow you to verify (and
confirm?) some of the statements made in the paper?
(iv)
Does the
information provided in the paper (e.g., tables) allow you to reproduce the
trend(s) shown in some of the figures?
(v)
Can you make
reasonable assumptions that will allow you to reproduce the trend(s) shown in
some of the figures?
(vi)
Do(es) the
conclusion(s) of the authors follow directly from the results and discussion
presented, and do(es) it/they make thermodynamic sense?
Summary of key concepts/issues in electrochemical thermodynamics:
-electric (electrostatic) potential ("electric pressure"): electric
energy/electric charge (f
= Ee/Q; 1 V = 1 J/1 C).
-electrochemical potential: m'i= mi + zFf (For z=0, uncharged species, it reduces to the chemical potential.)
-electromotive force (E):
It's NOT a force! But it IS a driving force, because it is a potential
difference... See Illustration 7.9 in Atkins8.)
-S/L adsorption equilibrium
can be severely affected by the development of a double
layer at the interface: in addition to dispersive attraction, need to
take into account the electrostatic repulsion or
attraction between the charged (electrode) surface and anions/cations in
solution.
-standard cell potential
(standard emf): Eo = -DGo/zF.
-galvanic cell: 'produces'
electric energy (i.e., converts chemical energy to electricity).
-electrolytic cell:
'consumes' electric energy (i.e., converts electric energy to, say, chemical
energy).
-Nernst equation (for a cell at equilibrium): ln K =
zFEo/RT. (If cell is NOT at equilibrium, then eq 7.29 in Atkins8
applies, where instead of K need to use the ratio of actual activities, and not
their equilibrium values.)
-Debye-Hückel equation (limiting law): -log g = Abs(z+z-)AI0.5 (i.e., it allows us
to determine activity coefficients, and thus activities, of ions in dilute
solutions of known ionic strength I; A=0.509 at 25 oC in aqueous
solution).
-Extended D-H eq:
introduces additional temperature-dependent parameter(s), see eq 5.72 in
Atkins8, as well as the effective ion diameter.
-electrochemical (redox) series
of metals: Au > Pt > Ag > ... > Ni > Fe > Zn > ... > Na
> Ca > K (e.g., Zn + AgO --> ZnO + Ag, Eo(Ag) > Eo(Zn)
=> "low reduces high".
Here is
Table 7.2 from Atkins8. (Consistent with electrochemical redox series of
metals? Should it be?)
HW3 (due
midnight 10/19, in Angel dropbox): E4.5, E4.9, P4.6, E5.18,
E5.20, E5.21, P5.6, P5.16, E6.11, E6.14b, E7.14, E7.16, P7.12, P7.17,
D16.1-16.3, D16.5, D16.6, E16.2, E16.8, P16.12, E17.1, E17.3, E17.10, P17.10.
-E4.5a: Tfr = 281.7 K. (Of course, as P
increases, so does the freezing point... But is this what makes ice skating
possible?)
-E4.5b:
Do we need to know the molecular weight of “a certain liquid”?
-E4.9a: Tfus = 272.7 K. (As expected?)
-P4.6: Tfr = 234.4 K. (As expected?)
-P5.6: At low solute concentrations its apparent MolWt is roughly
half of the actual (e.g., 26.8 g/mol at a molality of 0.037), so KF is totally
dissociated; at the higher concentrations, dissociation is NOT complete because
the apparent MolWt of KF is higher, 35-48 g/mol, and the ratio of molalities is
increasingly less than two. (Verified the Emsley paper?)
-P5.16: Is your graph similar to this one?
-E5.20: Why is the
"mean ionic activity coefficient" apparently 'based' (see Solution
Manual) on the component present in smaller amount?
-E6.11a: Tm is approx. 180 oC and xB
is approx. 0.26. The lower-right region is solid B+D, the middle region is D+L
(with D being the decomposing product of composition ABx, with
x=??), and so on.
-E6.14b: Surely we can find the UF4-ZrF4
phase diagram in the literature, and verify all the relevant statements...
Right?
-E7.16a: Do you agree that the activity coefficients of KBr
and Cd(NO3)2 are 0.77 and 0.67, and that therefore the
cell potential is -0.62 V?
-P7.12: Can you derive the expression DH = 2.3RT? And then, do you agree that the values for H2 and
CO are 14.7 and 18.8 kJ/mol? (Do these values make sense?)
-E16.2a: At 300 K it’s 2.57x1027 and at 600 K
it’s 7.26x1027 (units?).
-E16.8a: Molar energy and entropy at 10 K are 22 J/mol and
4.8 J/mol/K.
-P16.12: (a) (Incalculable?) exp(1.25x1024); (b)
(Incalculable?) exp(1.31x1024); (c) If W in (b) is greater than W in
(a), then...
-E17.1a: For I2 it’s 3.5R, for methane 3R and
for benzene 7R... Close enough to experimental values?
-E17.3a: At 25 oC it’s 19.6 and at 250 oC
it’s 34.3.
-E17.10a: rotational = -13.8 kJ/mol; vibrational = -0.20
kJ/mol.
-P17.10: By analogy with P17.11 (see Solutions Manual), the
ratios of translational and rotational partition functions are, respectively,
0.964 and 6.24. After some (manageable?) math, at 27 oC K=945; and
at 827 oC K=37. Obviously, you want to do this math “by hand” only
once, to see clearly how it works; subsequently, you want to use high-power software such as Gaussian or
Gamess (see below) or, at least, a template such as this
Mathematica file provided by Prof. Metiu with his recent PChem textbook.
Note: To find the (necessary?) values of rotational
constants for, e.g., methane, use Web of Science with “methane and rotational
constant*” to locate, for example, the 1994 paper by Hollenstein et al. (see
Table IV). Is there also a compilatory monograph of spectroscopic (including
rotational) constants of substances?
Note:
Developing a feel for RELATIVE values of fugacity
coefficients and activity
coefficients is among the MOST valuable claims to fame of an 'expert'.
Contrary to "popular belief", engineers, certainly -- and scientists
to a large extent -- are increasingly being paid to make reasonable estimates
and
assumptions,
and not so much to calculate...
Here
is an illustration of this process for activity coefficients. (Can you solve
the problem?) How would you update your information on the activity coefficients of electrolytes?
Pending conceptual questions:
-Is the characteristic shape
of the fugacity coefficient vs. pressure, or activity coefficient vs.
concentration, related in any way to (e.g., conditioned by) the
intermolecular potential vs. distance relationship (e.g., Lennard-Jones 6-12
curve)?
-Why is molality (mol/kg
solvent) a more convenient measure of concentration than molarity (mol/L
solution), or mole fraction, when dealing with electrolyte solutions? (Does the
addition of a second solute change the molality of the first? Is molality
temperature-dependent? Is density information needed?)
-Can you confirm the value
of A=0.509 kg0.5/mol0.5 in the Debye-Hückel eq?
-Can you confirm the
validity of this
table that conveniently summarizes the values of the mean ionic molality for common
types of electrolytes?
Exam
#1: Analyze the following paper: Anal Chem 2006,
78, 4642-53; see questions (i)-(vi) above. Due in Angel dropbox midnight
10/15.
-What is the meaning of a linear
van't Hoff plot? (For a typical homogeneous reaction -- e.g., CO+H2O=CO2+H2
-- is the van't Hoff plot linear over a wide temperature range?)
-Can you 'reproduce' Figures
2 and 3 using the information provided elsewhere in the paper? (Table(s)?)
-Which van't Hoff plots make
more sense, those in Figure 5 or those in Figure 7?
-The authors do not seem to
be sufficiently familiar with the Le Chatelier principle!? Are you surprised to
read the following: "It is well known that retention factors almost always
decrease with increasing temperature. The questions that we want to answer are,
Why? What does cause this decrease? How do the saturation capacities and the
equilibrium constants of adsorbates change with increasing temperature?"
Aren't the answers to at least two of these questions also well known?
-Do the authors provide any
justification for the existence of only two or three types of sites on the
heterogeneous adsorbent surface? Why not four or five? Or a continuous
distribution? With two or three, and without a physical interpretation, aren't
they just adding an excesive number of adjustable (and meaningless?!)
parameters to their model?
-Does the annotated paper
(see above) help to ease its 'digestion' and (critical) analysis?
-Do some of your van't Hoff
plots look like this
one?
-Is this Mathematica
template helpful for your nonlinear regression analysis? (Is a convenient
Excel template available as well? Such as this one?)
Summary of key concepts in statistical thermodynamics:
-quantum mechanics
<--> relativity: large Hadron collider ("standard model" OK?)
-quantum mechanics
<--> ('macrosopic') thermodynamics: statistical ('microscopic') thermodynamics
-Boltzmann distribution
(e.g., barometric pressure P=Poexp[-gMh/RT], or Maxwell's
distribution of molecular speeds)
-partition function (e.g.,
at T=0 only the ground state is accessible to a molecule, whereas at high T
virtually all energy levels are accessible)
-distinguishable
particles: Q=qN; indistinguishable
particles: Q=qN/N! (Stirling: lnX! = X lnX
- X)
-etc.
Here is a
nice example of the (fundamental and elegant!) link between quantum chemistry,
(statistical) thermodynamics and chemical kinetics.
--OVERVIEW AND APPLICATIONS OF
(CHEMICAL) KINETICS--
‘Mystery’ graphs: A
picture is worth “a thousand words”. A graph is worth much more, because it
summarizes and illustrates many scientific or engineering concepts; you ought
to think about a graph every time you walk into a shower… (What else are you
going to think about while you are there?!)
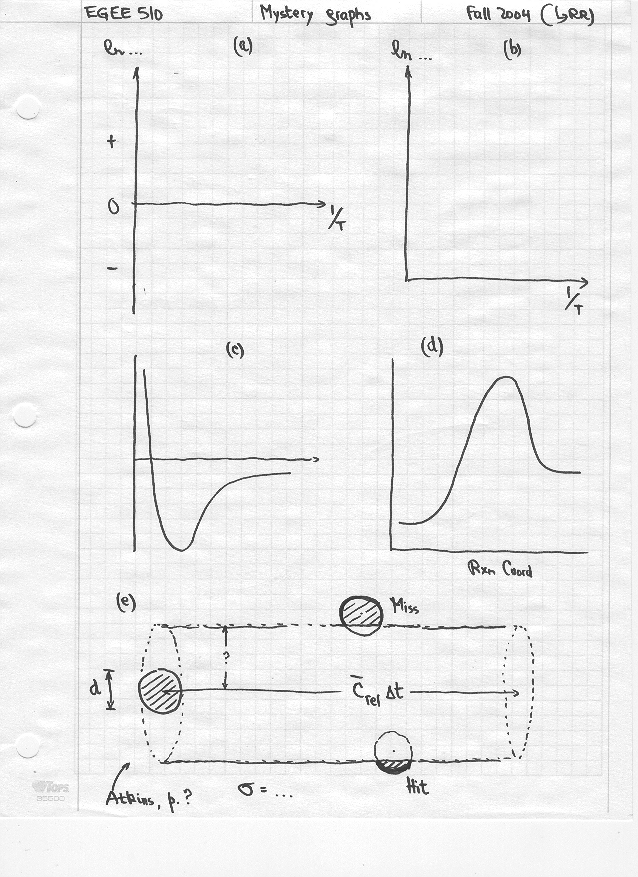{kind=link}
Below is a summary of key
issues/equations in relevant chapters in Atkins. (Be sure to work through the
units, verify their consistency, and develop a feel for the relevant orders of
magnitude. Here is a
Mathematica program which should help to give us that ‘feel’ for the key
parameters in the various equations.)
Key concepts:
-reaction rate, molecularity vs. order
-mean free path, collision diameter, collision frequency, c vs. crel
(kinetic theory of gases)
-collision theory
-Arrhenius equation (frequency factor, activation energy)
-transport coefficients (‘physical’ kinetics)
-steady-state approximation (SSA)
-transition-state theory
-liquid-phase concentrations
-any missing (that are important for your research?)
HW4 (due in Angel dropbox by
midnight 11/9; apart from these Atkins8 problems, see also Exercises 1-7
below):
-E21.2b: 475 m/s, 4.5x104 m, 1.1x10-2
1/s.
-E21.3a: Assuming lambda=0.1 m, 8.0x10-7 atm.
-E21.7a: 0.009.
-E21.14a: 16.9 J/s, 17 W (assuming 100% efficiency… Good
assumption? And if window is a fancy one, argon-filled, instead of air?)
-E21.22a: 7.6x10-3 S m2 mol.
-E21.29a: 0.42 nm.
-P21.16: Li, 0.237 nm
(vs. 0.059); Na, 0.184 (vs. 0.10); K, 0.125 (vs. 0.14);
Rb, 0.120 (vs. 0.15)… Comments? (These
‘subtle’ differences between Na+ and K+ in particular –
with and without their hydration shells – are important, of course, not only
for EGEE, but for life
itself, because of “ion channels” in cell membranes. As Eric Kandel
discusses in In Search of Memory,
2006, p. 89, “the ionic hypothesis … did for the cell biology of neurons what
the structure of DNA did for the rest of biology.”)
-P21.22: 0.83 nm (with u=1.1x10-4 cm2/s/V).
-E24.2b:
-E24.4a: 1.03x10-5
m3/mol/s.
-E24.7a: 7.4x106 m3/mol/s,
1.3x10-7 s.
-E24.14a: -45.8 J/mol/K, 5.0 kJ/mol, 18.7 kJ/mol.
-E24.15a: 20.9 L2/mol2/min.
-P24.2: P=0.0065, and the reactive cross-section is 3.9x10-21
m2. (Temperature-dependent?)
-P24.4: The second point in the table should be -20.93 oC,
instead of -20.73… Right? Therefore, with Ea=86 kJ/mol and A=1.26x1014
s-1, DH=84 kJ/mol (makes sense?), DS=18 J/mol/K (makes sense for a thermolysis reaction?), DU=84 kJ/mol, and DG=79 kJ/mol.
-P24.10: DH=60 kJ/mol, DU=63 kJ/mol, DS=-181 J/mol/K, DG=115 kJ/mol. (See
Note 4 below!)
-P22.2: After 43.8 h the concentration drops to ca. 0.97
mol/m3.
-P22.10: Do you agree that k=7.6x10-4 1/s?
-P22.16: Do your results confirm those of the original
paper (3.98 kcal/mol and 11.1x109 L/mol/s)?
-P23.1: The appropriate time interval, for the
concentrations and k values as shown, is NOT 0-10 ns (as in the problem statement)
but 0-10 ms (as shown in the solution manual). Do you agree? (Note that, as
expected for a consecutive reaction scheme, there is a more or less monotonic
decrease in the concentration of H and NO2, O and OH are
intermediate products whose concentration therefore goes through a maximum,
whereas the appearance of the final product O2 is ‘delayed’.
-P23.2: The most appropriate kinetics is
pseudo-first-order… Right? (But does it reproduce properly the experimental [O]
vs. t behavior?)
Notes:
-if you do not agree with the answers to the HW questions
that will eventually be posted here (before the due date of the HW), let’s
discuss them. (The procedures used are, of course, more important than the
exact numbers obtained…)
-in E21.14 it is asumed that the RDS is heat transfer
through the gas space between the panes, and NOT through either of the two
glass panes; is this a reasonable assumption? (Are you familiar with the
concept of R-value of insulating materials? See what you can find about it on
wikipedia or using google.com. Also, when you walk toward campus from the
east-side corner of College and Burrowes, be sure to read this!)
{kind=link}
-in analyzing the units of the various quantities involved in
the motion of ions (in liquids), remember that 1 volt is 1 joule per coulomb (1
V = 1 J/C).
-see here a
clarification of the relationship between the observable (experimental) activation
energy and the internal energy increase required for the formation of an
activated compex (transition state): in analogous fashion, one can derive the relevant
expressions for a unimolecular reaction, and then conclude that the entropy
of activation in E24.14b should be -32.4 and not -24.1 J/mol/K. (Do you agree?)
Applications where these issues are important are, for example:
-combustion of gases and liquids (homogeneous kinetics)
-atmospheric reactions (e.g., soot + ozone = CO + O2)
-aqueous-phase reactions (e.g., SO2(aq) -> SO3(aq)
-> H2SO4(aq))
-thermal decomposition of organic compounds (e.g.,
pyrolysis of oil shale, biomass and coal, decomposition of soil constituents)
-gasification and combustion of solids (heterogeneous
kinetics, catalysis)
-others? (Here list some of YOUR topics of interest…)
One of the most important
skills to be developed is the “feel” for orders of magnitude (see class
handout). Toward this goal, it’s always instructive to put some important
markers on the scale of chemical reaction rates:
(a) A very fast rate: Show (using a class handout) that a
typical coal (carbon) combustion rate is of the order of 1 mol/cm3/s.
(Gas-phase combustion reactions, as we saw, are some three orders of magnitude
faster than this.)
(b) At the other extreme, let’s show that the rate of the
metabolic process(es) in our body is some 10 orders of magnitude less than
that.
The
“bottom line” of this section, for our purposes, is the following:
-using the concepts of mean free path
and mean molecular speed, developed from the Maxwell distribution
function, can quantify collisions between molecules, or of molecules with
surfaces, and thus estimate the preexponential term in the chemical kinetic
rate expression
-using the same concepts, can estimate
transport coefficients
-in conjunction with the
Stokes-Einstein and Einstein-Smoluchowski equations, establish a link between
microscopic parameters of particle motion (e.g., mean free path, random walk
step) and macroscopic (observable) properties of fluids
(This
analysis can then by followed up by statistical thermodynamics, or statistical
mechanics, and then quantum mechanics, all of which increasingly allow the
calculation of rates of both physical and chemical processes from “first
principles”… but this is, of course, beyond the scope of our discussion.)
Collision frequency/flux (from kinetic theory of gases):
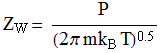
This equation leads to the collision
theory of chemical kinetics, whose key equations are the following:
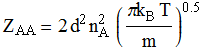
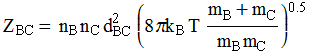
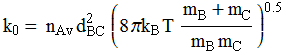
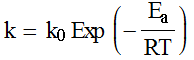
![]()
Related (and more basic) equations are those for the mean molecular speed, mean
free path, and the diffusivity (of an ideal gas):
![]()
![]()
![]()
Exercise 1. A recent physical chemistry textbook (Silbey et al., 2005) reports the following values for the interaction between O2 and H2 molecules at 25 oC.
(a)
Collision frequency of H2 with O2 molecule = 4.63x109
s-1;
(b)
Collision frequency of O2 with H2 molecule = 9.258x109
s-1;
(c)
Collision density between H2 and O2 molecules = 1.244x108
mol/L/s.
Verify them! [The complete
text of the problem statement is as follows (p. 638): “A gas mixture contains H2
at 0.666 bar and O2 at 0.333 bar at 25 oC. (a) What is
the collision frequency z12 of a hydrogen molecule with an oxygen
molecule? (b) What is the collision frequency z21 of an oxygen
molecule with a hydrogen molecule? (c) What is the collision density Z12
between hydrogen molecules and oxygen molecules in mol L-1 s-1?
The collision diameters of H2 and O2 are 0.272 nm and
0.360 nm, respectively.”]
Exercise 1a. See here the Mathematica
solution to Example 24.1 in Atkins. Here you can see,
as in Exercise 2 below, that the
fraction of effective collisions is small, of the order of 10-6.
Exercise 1b (McQuarrie and Simon, Physical Chemistry, Ch. 27).
Consider a mixture of methane and nitrogen in a 10 L container at 300 K with
partial pressures of 65 and 30 mbar, respectively. Show whether the collision
frequency of a methane molecule with N2 molecules is indeed 2.8x108
s-1, and the frequency of CH4/N2 collisions is
4.4x1032 molecules/m3/s.
Here is a
template for numerical exercise #21.15 in Atkins…
Exercise 2. Calculate the number of collisions of methane
molecules with O2, in moles/cm3/s, assume zero-order
reaction and estimate the time required for complete consumption of methane,
assuming that all collisions are ‘effective’. Based on the order of magnitude
obtained for this time, discuss whether the latter assumption is reasonable.
(Hint: Combustion of natural gas is fast, but not that fast! The fraction of
effective collisions turns out to be ca. 1/106.)
Exercise 3. A typical value for the collision frequency
(which in some cases can be equated with the preexponential factor in
the Arrhenius rate expression) is 1013 s-1. The following
experimental results confirm this for the decomposition reaction 2N2O5
-> 2N2O4 + O2. Determine the activation
energy and the preexponential factor.
(What is the order of most decomposition reactions? Is this fact
consistent with the law of mass action? See the handout on the
Lindemann-Hinshelwood mechanism.)
T (K) 288 298 313 323 338
k x 105 (s-1) 1.04 3.38 24.7 75.9 487
Silbey et al. (Physical
Chemistry, 4th edition) report the following results for the
reaction N2O5 --> 2NO2 + 0.5O2:
T (K) 273 298 308 318 328 338
k x 105 (s-1) 0.0787 3.46 13.5 49.8 150 487
Is this consistent with the
previous results?
McQuarrie and Simon (Physical
Chemistry, 1997, p. 1146) report the following results for the same reaction at
318 K. Do they agree with the previous results?
t/min 0 10 20 30 40 50 60 70 80 90 100
[N2O5]/10-2
mol dm-3 1.24 0.92 0.68 0.50 0.37 0.28 0.20 0.15 0.11 0.08 0.06
Exercise 4. Froment and Bischoff (1990 CRE textbook) report the
following kinetic data for thermal cracking (pyrolysis, decomposition) of
ethane:
T (oC) 702 725 734 754 773 789 803 810 827 837
k (s-1) 0.15 0.27 0.33 0.59 0.92 1.49 2.14 2.72 4.14 4.67
(a) Determine the activation
energy and the preexponential term and comment whether these values are
‘reasonable’. (See also Exercise #5 below.)
(b) Use the Web of Science to
find out whether there is agreement in the literature about these values.
An alternative theory to that of
“effective collisions” is the transition state theory.
Let’s now summarize its key equations.
Van’t Hoff has provided an elegant,
thermodynamic interpretation of the temperature dependence of the reaction
rate. (Of course, the name of Arrhenius is associated with this equation; he
verified it experimentally.) Let’s start with the Law of Mass Action (Guldberg
and Waage, 1864):
aA + bB + ... F pP + qQ + ...
![]()
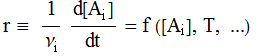

![]()

At
equilibrium, by definition,
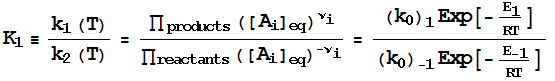
Therefore,
since
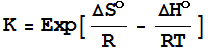
we
have a neat link between kinetics and thermodynamics
(at least in principle):
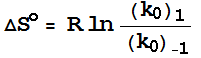
![]()
![]()
Remember
one of our ‘mystery’ graphs!
It
is now intuitively obvious (and can be easily derived; see Atkins) that the rate
constant from transition state theory (or activated
complex theory) can be obtained using the Eyring equation (see handout
on Reactivity):
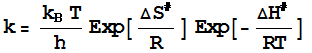
You can now show that the
preexponential factor, or the frequency factor here, is indeed typically of the
order of 1013 s-1.
Using
quantum chemistry methods similar to, but more sophisticated than, the HMO
method, the geometries and energies of
transitions states can be obtained by solving the Schrödinger equation,
and thus the activation energies and the rate constants can be estimated for an
increasing number of practically interesting reactions.
Here is a
bottom-line summary of the kinetics of liquid-phase rxns.
Here is a brief
empirical (macroscopic) summary on diffusion…
For
a fundamental microscopic view, a rewarding exercise is to reproduce the
calculation of mean free path that Einstein made in his “miraculous-year”
Brownian motion paper? For those of you who like to read only the “original
stuff”, here
it is, courtesy of google.com (and of
And here is a
brief summary on chain reactions. Very profitable reading are Hinshelwood’s
and Semenov’s
Nobel lectures given in 1956.
Perhaps the most important concept in
the whole of science and engineering is that of the RDS. Let’s first draw the
instructive analogy between electric circuits (remember Ohm’s law?) and
consecutive chemical reactions. And then let’s show how RDS makes
easier our analysis of the most important chemical reaction in the world
(and in EGEE): nC + [(n+1)/2]O2 = CO2 + (n-1)CO.
Let’s analyze a practical example: the
difference between pulverized coal combustion (small particles, high T) and
fluidized-bed combustion (larger particles, lower T). Study this Mathematica file
carefully, so that you can get a feel for the importance of the key parameters.
Another key concept in this field is
that of a chain
reaction, with its initiation, propagation
and termination steps. A related concept, which
allows us to digest more easily such complex reactions, is SSA. Let’s analyze a representative
reaction, ethane decomposition, using the Rice-Herzfeld
mechanism (see also relevant class handout):
(1) C2H6 à 2CH3 k1
(2) CH3 + C2H6
à CH4 + C2H5 k2
(3) C2H5 à C2H4 + H k3
(4) H + C2H6 à H2 + C2H5 k4
(5a) H + C2H5 à C2H6 k5
Of
course, the math becomes quite involved when many such reactions must be
considered (hundred or more elementary reactions are not uncommon), and
software such as ChemKin
becomes indispensable even for routine analyses (e.g., of concentration vs.
time profiles). For simpler problems, such as the one shown here, a home-made program
will suffice.
Exercise 5. Show that the reaction rate is indeed first-order!
Instead of reaction (5) shown above, assume the following termination reaction:
(5b)
2C2H5 à C4H10 k5 = 4.0x108 Exp[-0/RT].
(a) Use the provided
Mathematica template to recalculate the relevant concentration profiles. Which
are the dominant products?
(b) Can you compare the
results in Exercise 4 with the ones obtained here?
For
example, verify that the SSA results in the following expression for the rate
of decomposition of ethane: (2k1 + k3(k1/k5)0.5)
[C2H6].
Note: The
kinetic expression for eq. 3 is k3 [C2H5] [C2H6]0.5
instead of the straightforward k3 [C2H5]. Can
you find out why (say, using the Web of Science)?
Now, reducing this expression, as well as the one
using eq. 5b instead of eq. 5a, to a first-order equation, are the rate
constants of the same order of magnitude?
(c) Does methane formation
become more important at higher temperatures?
Here is an Excel file
that compares the results of a mechanistic analysis of ethane decomposition
(Rice-Herzfeld) with those of a phenomenological analysis (Exercise 4). Be sure
to verify whether it is correct!
A nice example of a
real-world situation in which the virtues of mixed flow and plug flow must be
combined is the combustion
chamber (e.g., in a power plant or an automobile engine). This is
best understood by ‘playing’ with the parameters of the kinetic equation that
describes an autocatalytic reaction (e.g., A + P
à P + P), and
reproducing the characteristic maximum in the rate vs. conversion behavior. Here is a template
that you can use as a starting point in this exercise.
With this discussion we can
now appreciate the fact that chemical kinetics applied to EGEE problems spans
the (very wide but digestible and elegant) range from
the Schrödinger equation -- e.g., the reactivity of sites in aromatic
molecules or the geometry of the transition state (remember also the term kBT/h
in transition-state theory…) -- to the reactor
performance (design) equation.
HETEROGENEOUS AND CATALYTIC REACTION KINETICS
-Here is a summary
of the principal heterogeneous reaction/catalysis
issues
-see also Ch. 25 in Atkins_8
In EGEE applications, many
chemical reactions are heterogeneous, i.e., with reactants in different phases
(e.g., gas/solid), and many are also catalyzed and are thus heterogeneous. In
catalytic reactions, the rate most often increases as the catalyst particle
size decreases (because catalyst dispersion
thus increases).
Exercise 6. For a cubic solid with 5 faces exposed to an
adsorbate (or a reactant), verify that catalyst dispersion (D, or fraction
exposed) can be calculated using the following expression: five times the molecular
(or atomic) weight divided by the product of particle diameter (d), density,
molecular (or atomic) cross-section and Avogadro number. Show that, in many
cases, this approximately reduces to D = 1/d, where d is in nm. (Note: D is
often determined from chemisorption experiments, and d is most often determined
from TEM experiments.)
Exercise 7. A TEM analysis of a carbon-supported CaO catalyst
(with 10% CaO by weight) shows particles whose average diameter is 30 nm. (a)
Determine the surface area and the dispersion (fraction exposed) of this
catalyst. (b) What would be the chemisorption uptake of CO2 (in
cc(STP)/g catalyst) if the adsorption stoichiometry is 1/1?
For a summary of vessels (“reactors”) in which chemical reactions take place,
see here.
The objective here, of course and as appropriate in an engineering
class, is to show the direct impact of chemical
kinetics on reactor size and therefore on process economics.
Exam #2 (due in Angel dropbox by midnight
11/16): Let’s ‘dissect’ the classical
paper(s) by Daniels
and coworkers on the decomposition of N2O5: JACS 43,
53 (1921); 52, 1472 (1930). Many PChem textbooks also contain an
analysis of this “first homogeneous first-order reaction to be investigated”
(Laidler, in “Chemical Kinetics,” 1950, p. 219).
-Figs. 2 and 3 show the ‘raw’ data.
-Figs. 4 and 5 and Tables I-V show data analysis. Select at
least one point in Figure 4 and explain how it was obtained from the raw data
and the equation(s) presented in the text. Do the same in Table I by showing
the sequence of calculations for a representative row; for example, at 25 oC
and PO2=5 torr, 1/(8KPO2)=2.43, then a=0.761, 3PO2 +2aPO2=22.6,
etc. As a reminder of your thermo expertise, explain why K decreases as T
increases (p. 58; can you verify the validity of the equation used?). In Table
II we see, for example, from Figure 2 that, indeed, at 25 oC and
1200 min, k=2.303/(1200-20) log[(288.7-23.9)/(288.7-268.7)]=0.0022 min-1.
Right? Another example?
-Table VI shows a typical kinetic correlation (calculation
of activation energy and pre-exponential factor). Note, of course, that one of
the k values for 25 oC should be 0.00201, and not 0.00801. (Are
there any other typos or errors?) Do the k values in this table agree with
those shown in Exercise 3 above?
-In addition to answering and commenting on the questions
listed above, be sure to address the same six general questions that we had on
Exam #1. (Of course, where it says ‘thermodynamics’ there, ‘kinetics’ applies
here.)
-Select at least one citing reference and discuss the
context (Appropriate? Too general? Specific enough?) in which the Daniels and Johnston
paper is cited by the citing author(s).
End-of-semester activity (no class
meetings on 11/19, 12/8 and 12/10):
(1) Let’s get a suitable (and affordable!) software
(Gamess?) for doing quantum chemistry
calculations (see Part 2 in Atkins8, especially Chs. 8-13).
(2) Let’s reproduce a suitable kinetics tutorial. (Remember
the Ochterski example?)
(3) Let’s use this program to determine the rate constant
of a suitable
and relevant reaction (Exam #3?).
-For a hands-on introduction
to Gamess, see, for example, http://www.msi.umn.edu/tutorial/chemistryphysics/Intro_to_GAMESS.pdf.
We are going to be interested primarily in determining vibrational frequencies
and -- as a follow-up (see the “Ochterski_2000.pdf” summary mentioned above) --
in thermochemical properties (of the reactants and of the activated complex,
whose geometry has been optimized previously). And for the most authoritative
introduction to Gamess, see http://www.msg.chem.iastate.edu/tutorials/gamessintro.pdf,
especially p16.
-ChemViz
is a convenient (and affordable!) tool for visualizing and analyzing the
geometry of molecules.
-For generation of Z-matrix
(necessary for Gamess input file) and its conversion to cartesian coordinates,
see http://www.shodor.org/chemviz.
Here is a typical input file for the
calculation of energy of a molecule, as well as the corresponding output file. Can you
modify/adapt them to include the calculation of thermodynamic properties? (Have
you been able to find similar or more appropriate ‘templates’? For example, at
the San Diego Supercomputer Center, http://education.sdsc.edu/download/chemistry/gamess_logfile_tutorial_ex3.htm?
And perhaps the tutorial “Studying
Chemical Reactions with PC GAMESS” is useful as well…)
A judicious search within the
Web
of Knowledge can halp you identify a reaction of (your) interest for
which much information is already available regarding the geometry of the
transition state… Here are some examples:
-“transition state” (title) and “ab initio” (title)
-Gamess (topic)
-Gamess (topic) and “transition state” (title)
-Gamess (topic) and “vibrational frequenc*” (topic)
-Others? (e.g., Gamess (topic) and “[your favorite
molecule]”)
Exam #3: due 12/19 (in Angel dropbox), at the latest.
(a) Submit your results of
“End-of-semester activity” (see above), OR
(b) Use quantum chemistry
software (Gamess?) to solve P11.18a and confirm the results of E11.5a and
E11.5b in Atkins8. (Note: If Gamess is inconvenient to use, are the links
provided by Atkins8 more user-friendly?)
LRR3@psu.edu (updated 12/03/2008)